
Стандартное лечение ВИЧ-1-инфицированных пациентов, как правило, предусматривает применение трехкомпонентной схемы антиретровирусной терапии (АРТ) в режимах, включающих 2 НИОТ и третий препарат, который представляет собой ННИОТ, усиленный ИП (уИП), либо ИИ. Последние достижения в области АРТ ВИЧ-инфекции и углубленное понимание проблемы устойчивости ВИЧ к лекарственным препаратам позволили рассмотреть возможность переключения пациентов с вирусологически эффективных схем терапии на альтернативные режимы в определенных ситуациях [1, 2]. Например, некоторые препараты группы НИОТ могут вызывать анемию (зидовудин – ZDV) [3], нейротоксичность и липодистрофию (ставудин – d4T и диданозин – ddI) [4], а также почечную дисфункцию (тенофовир – TDF) [5]. В ряде когортных исследований были зарегистрированы случаи развития сердечно-сосудистых заболеваний на фоне приема абакавира (ABC) [6].
Согласно российским и международным клиническим рекомендациям, возможен перевод пациентов на двойные режимы терапии. Основной принцип переключения заключается в поддержании вирусологической супрессии. Кроме того, сохраняется возможность применения других вариантов лечения в будущем. Показаниями для смены режима терапии на фоне вирусологической супрессии могут быть уменьшение количества принимаемых препаратов и кратности их приема, улучшение переносимости лечения и снижение кратковременной и долговременной токсичности, предотвращение или снижение выраженности лекарственных взаимодействий, оптимизация АРТ в связи с беременностью или ее планированием, снижение стоимости лечения, а также ряд других причин [2].
Появляется все больше данных о том, что схема терапии, включающая уИП + ламивудин (3ТС), может поддерживать вирусологическую супрессию у пациентов без первичных мутаций резистентности, ранее не получавших АРТ [7], а также у пациентов с устойчивой вирусологической супрессией [8]. 3ТС зарекомендовал себя как эффективный препарат с хорошим профилем безопасности, который широко применяется в терапии первой линии у ВИЧ-1-инфицированных взрослых и детей. Резистентность к нему возникает при наличии единственной точечной мутации в геноме ВИЧ-1 (мутация гена обратной транскриптазы M184V), при которой препарат теряет эффективность, но которая, в свою очередь, отрицательно влияет на способность вируса к репликации. Комбинированный препарат лопинавир/ритонавир (LPV/r) продемонстрировал выраженную противовирусную активность на широкой популяции пациентов в ходе разных клинических исследований продолжительностью до 7 лет [9].
Применению двухкомпонентной схемы LPV/r + 3TC у ВИЧ-1-инфицированных пациентов посвящены исследования GARDEL и OLE.
В международном исследовании GARDEL [7] изучали результаты лечения в течение 48 нед. взрослых пациентов (в возрасте ≥ 18 лет), ранее не получавших АРТ, с зарегистрированной ВН ВИЧ-1 не менее 1000 копий/мл. Пациенты были рандомизированы (1:1) в группы двух- или трехкомпонентной терапии. Двухкомпонентная схема включала прием LPV/r 400/100 мг + 3TC 150 мг 2 раза в сутки. Трехкомпонентная схема включала LPV/r 400/100 мг 2 раза в сутки и 3TC или эмтрицитабин (FТC) + другой НИОТ в виде комбинации с фиксированными дозами. Первичной конечной точкой являлась частота вирусологического ответа через 48 нед. АРТ. Граница не меньшей эффективности для разницы между группами составляла -12%. 198 пациентов в группе двухкомпонентного и 175 пациентов в группе трехкомпонентного режима завершили 48-недельный курс лечения. Различия в частоте ответа у пациентов двух групп на 48-й неделе терапии составили 4,6% (95% ДИ -2,2–11,8; p = 0,171). У пациентов с исходной ВН не менее 100 000 копий/мл были получены схожие результаты: 87,2 и 77,9% соответственно, разница составила 9,3% (95% ДИ -2,8–21,5; p = 0,145). В связи с токсичностью или непереносимостью прекратили лечение 10 (4,9%) пациентов в группе трехкомпонентной терапии и только 1 (0,4%) пациент в группе двухкомпонентной терапии. Разница составила 4,5% (95% ДИ -8,1– -0,9; p = 0,011).
В рандомизированном открытом исследовании OLE [8] 250 ВИЧ-инфицированных взрослых пациентов с уровнем РНК ВИЧ-1 < 50 копий/мл в течение не менее 6 мес. на фоне трехкомпонентной терапии по схеме LPV/r (2 раза в сутки) + 3TC или FТC и другой НИОТ без вирусологической неудачи или резистентности к данным препаратам были рандомизированы (1:1) в 2 группы для продолжения АРТ по трехкомпонентной схеме или переключения на двухкомпонентную схему (LPV/r 400/100 мг перорально 2 раза в сутки + 3TC 300 мг перорально 1 раз в сутки). Первичной конечной точкой являлся ответ на лечение через 48 нед. Граница не меньшей эффективности составляла -12%. Анализ результатов в популяции intention-to-treat показал, что ВН сохранялась на уровне < 50 копий/мл у 110 (86,6%) пациентов из 127 в группе трехкомпонентной терапии и у 108 (87,8%) пациентов из 123 в группе двухкомпонентной терапии. Разница составила -1,2% (95% ДИ -9,6 – 7,3; p = 0,92), что удовлетворяло критериям не меньшей эффективности. Частота развития серьезных нежелательных явлений (НЯ) в обеих группах была сопоставима: 8 (7%) и 5 (4%) соответственно (p = 0,515) [8].
 Таким образом, данные, полученные в международных исследованиях, показывают, что применение LPV/r + 3TC в качестве схемы первой линии у пациентов без опыта АРТ и поддерживающей терапии у пациентов с достигнутой вирусологической супрессией имеет не меньшую эффективность, сопоставимый или даже более благоприятный профиль безопасности и меньшую частоту прекращения терапии в связи с токсичностью или непереносимостью по сравнению с трехкомпонентной схемой LPV/r + 2 НИОТ. Преимуществом является и то, что упрощение режима АРТ может значительно снизить стоимость лечения, особенно учитывая, что 3TC является хорошо зарекомендовавшим себя воспроизведенным антиретровирусным препаратом.
Таким образом, данные, полученные в международных исследованиях, показывают, что применение LPV/r + 3TC в качестве схемы первой линии у пациентов без опыта АРТ и поддерживающей терапии у пациентов с достигнутой вирусологической супрессией имеет не меньшую эффективность, сопоставимый или даже более благоприятный профиль безопасности и меньшую частоту прекращения терапии в связи с токсичностью или непереносимостью по сравнению с трехкомпонентной схемой LPV/r + 2 НИОТ. Преимуществом является и то, что упрощение режима АРТ может значительно снизить стоимость лечения, особенно учитывая, что 3TC является хорошо зарекомендовавшим себя воспроизведенным антиретровирусным препаратом.
Бо́льшая часть данных об эффективности применения схемы LPV/r + 3TC у ВИЧ-1-инфицированных пациентов с предшествующим опытом лечения была получена в международном исследовании OLE. В связи с этим целью настоящего неинтервенционного продольного продукт-ориентированного многоцентрового исследования являлась оценка вирусологической эффективности двойной схемы (LPV/r + 3TC) у ВИЧ-1-инфицированных пациентов с предшествующим опытом АРТ и неопределяемым уровнем РНК ВИЧ-1 в плазме крови (в течение не менее 6 мес.) в рутинной клинической практике в Российской Федерации.
Материалы и методы
Дизайн исследования – проспективное неинтервенционное многоцентровое продольное продукт-ориентированное, без контрольной группы. Набор пациентов проводили в 13 федеральных и республиканских центрах по профилактике и борьбе со СПИД и инфекционными заболеваниями в 12 городах Российской Федерации (Москва, Санкт-Петербург, Калининград, Екатеринбург, Оренбург, Иркутск, Уфа, Челябинск, Красноярск, Казань, Ростов-на-Дону, Барнаул). Критериями отбора центров для исследования было наличие пациентов, подходящих для участия в исследовании, и готовность проводить его надлежащим образом. Исследование проведено в соответствии с Хельсинкской декларацией и соответствующими локальными регуляторными требованиями, одобрено Независимым междисциплинарным комитетом по этической экспертизе клинических исследований (Москва, Россия). Все пациенты предоставили письменное разрешение (согласие) на использование/раскрытие их данных до начала проведения процедур в рамках исследования.
Подходящими для участия в исследовании считались ВИЧ-инфицированные пациенты мужского и женского пола в возрасте 18 лет и старше с достигнутой вирусной супрессией (в 2 последовательных измерениях уровень РНК ВИЧ-1 в плазме крови < 50 копий/мл), сохраняющейся в течение, по крайней мере, 6 мес. на фоне любой трехкомпонентной схемы АРТ. Пациенты должны были иметь совокупный непрерывный опыт лечения антиретровирусными препаратами в течение не менее 6 мес., а также рекомендацию к переходу на режим LPV/r + 3TC по решению лечащего врача в условиях рутинной клинической практики. К участию также допускали пациентов, которые были переведены на режим LPV/r + 3TC более чем за 60 дней до включения в исследование. Критериями исключения из исследования являлись наличие противопоказаний к применению LPV/r и 3TC или предшествующее участие в данной программе.
 Пациентам были запланированы 5 наблюдательных визитов в рамках рутинной клинической практики и консультации лечащего врача приблизительно каждые 3 мес. Переключение с трехкомпонентной схемы на двухкомпонентную выполняли на Визите 1 (День 0). Пациенты получали АРТ по схеме LPV/r + 3TC в соответствии с предписаниями врача и утвержденной инструкцией по применению в суточных дозах 800/200 мг LPV/r (400/100 мг 2 раза в сутки) + 300 мг 3TC (150 мг 2 раза в сутки) в течение 48 нед. и находились под наблюдением в течение 30 дней после получения последней дозы препаратов в рамках исследования. Исследуемые препараты были коммерчески доступными.
Пациентам были запланированы 5 наблюдательных визитов в рамках рутинной клинической практики и консультации лечащего врача приблизительно каждые 3 мес. Переключение с трехкомпонентной схемы на двухкомпонентную выполняли на Визите 1 (День 0). Пациенты получали АРТ по схеме LPV/r + 3TC в соответствии с предписаниями врача и утвержденной инструкцией по применению в суточных дозах 800/200 мг LPV/r (400/100 мг 2 раза в сутки) + 300 мг 3TC (150 мг 2 раза в сутки) в течение 48 нед. и находились под наблюдением в течение 30 дней после получения последней дозы препаратов в рамках исследования. Исследуемые препараты были коммерчески доступными.
Первичной конечной точкой исследования являлась доля пациентов с неопределяемой ВН на фоне применения двойной схемы АТР на 48-й неделе исследования. На каждом визите собирали данные о ВН. Если РНК ВИЧ-1 определялась в плазме крови на одном из визитов, но не определялась на предшествующем визите, через месяц проводили повторное (контрольное) исследование. Если по его результатам концентрация РНК ВИЧ-1 в плазме крови составляла > 400 копий/мл, проводили исследование лекарственной устойчивости ВИЧ-1 методом генотипирования.
Вторичными конечными точками оценки эффективности являлись доля пациентов с вирусологической супрессией на 24-й неделе, ВН и количество CD4+-лимфоцитов на 24-й и 48-й неделях. Оценка безопасности включала анализ лекарственной устойчивости к каждому классу препаратов, оценку антропометрических показателей, биохимических показателей крови и частоты развития серьезных и несерьезных НЯ.
Сбор данных осуществляли в ходе бесед с пациентами, их осмотра и из первичной документации в исследовательском центре. Данные регистрировали в электронных индивидуальных регистрационных картах, которые заполняли на каждого пациента. При выявлении расхождений их доводили до сведения исследователя и устраняли.
Статистический анализ выполняли с использованием программного пакета SAS® версии 9.3. План статистического анализа был подготовлен и финализирован до закрытия базы данных. Для анализа и представления данных использовали методы описательной статистики. В рамках исследования формальной проверки гипотезы не проводили. Количественные переменные были представлены в сводном виде с вычислением следующих статистических показателей: число пациентов (n), среднее, стандартное отклонение (СО), медиана, 1-й и 3-й квартили, минимальное (min) и максимальное (max) значения. Качественные переменные были представлены в сводном виде с указанием числа и доли пациентов в каждой категории.
Качественные переменные эффективности и безопасности были представлены по категориям с построением двухстороннего 95% ДИ для одной доли простым асимптотическим методом. Количественные переменные (количество CD4+-лимфоцитов, ВН, лабораторные показатели и антропометрические характеристики) были представлены в сводном виде с расчетом статистических показателей и 95% ДИ для средних значений. НЯ были закодированы согласно терминологии Медицинского словаря для регуляторной деятельности (MedDRA) версии 18.1 и представлены по классам системы органов (КСО), предпочтительным терминам (ПТ) и частоте возникновения.
Размер выборки рассчитывали, основываясь на результатах исследования OLE, в котором у 87,8% пациентов с предшествующим опытом лечения достигнутая вирусная супрессия сохранялась на фоне терапии по схеме LPV/r + 3TC через 48 нед. наблюдения. Исходя из предположения, что в настоящем исследовании доля пациентов с неопределяемой ВН в плазме крови на 48-й неделе будет сопоставимой, размер выборки был определен равным 196 пациентам. Предполагалось, что половина ширины двухстороннего 95% ДИ, рассчитанного простым асимптотическим методом для одной пропорции при нормальном распределении, будет около 5% от наблюдаемой доли пациентов при ожидаемой доле 85%. С учетом возможного выбывания в ходе исследования 10% пациентов число пациентов для включения в исследование было определено равным 216.
 Результаты
Результаты
В период с ноября 2015 г. по май 2017 г. в исследование были включены 216 пациентов, соответствующих критериям отбора, в 13 центрах по борьбе со СПИД и инфекционными заболеваниями Российской Федерации. Из них 202 (93,52%) пациента завершили исследование по протоколу, 14 выбыли из исследования досрочно по разным причинам: 2 пациента – из-за неэффективности терапии, 3 пациентки – в связи с беременностью; 4 пациента отозвали согласие на участие; с 5 пациентами был утерян контакт. 7 человек прекратили участие в исследовании до Визита 2 (12-я неделя), 4 – до Визита 3 (24-я неделя), в том числе 2 пациента с документально подтвержденной неэффективностью терапии. С 1 пациентом до Визита 4 (36-я неделя) и 2 пациентами до Визита 5 (48-я неделя) была утеряна связь.
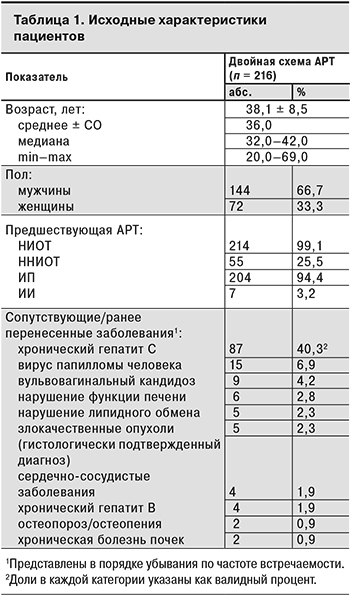
Исследуемую популяцию составили ВИЧ-инфицированные пациенты мужского и женского пола европеоидной расы в возрасте от 20 до 69 лет с предшествующим опытом АРТ и предварительно достигнутой супрессией РНК ВИЧ-1 в плазме крови (исходная ВН < 50 копий/мл). 99,1% пациентов до включения в исследование получали лечение по схемам, включающим НИОТ и уИП и/или ННИОТ, и/или ИИ (меньшее число пациентов).
У большинства пациентов не было клинически значимых сопутствующих заболеваний в анамнезе, однако 87 (40,3%) участников имели антитела к HCV. Менее чем у 10% пациентов в анамнезе были отмечены вирус папилломы человека, хронический гепатит B, нарушение функции печени (повышение функциональных проб печени > 5 верхней границы нормы), хроническая болезнь почек (клиренс креатинина менее 60 мл/мин в течение более чем 3 мес., пиелонефрит, гломерулонефрит), сердечно-сосудистые заболевания (документально подтвержденный инфаркт миокарда, стенокардия, гипертония, инсульт или необходимость медикаментозного лечения указанных состояний), нарушение липидного обмена (применение лекарственных препаратов для снижения повышенного уровня холестерина или триглицеридов – статины, эзетрол, фибраты, ниацин), вульвовагинальный кандидоз, остеопороз/остеопения и злокачественные опухоли (диагноз подтвержден гистологически). Исходные характеристики пациентов представлены в табл. 1.
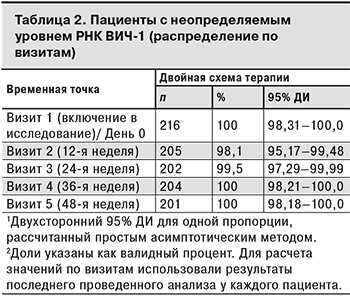
Данные о ВН были зарегистрированы у 209, 203, 204 и 201 пациента на 12-й, 24-й, 36-й и 48-й неделе терапии соответственно. Из 202 пациентов, завершивших исследование, у 1 пациентки такая информация отсутствовала на Визите 5. В табл. 2 представлены данные о пациентах с вирусологической супрессией по визитам исследования.
У 4 (1,9%) пациентов на Визите 2 ВН составляла ≥ 50 копий/мл, впоследствии у 2 из них определяемый уровень ВН был подтвержден результатами повторных тестов и составил > 400 копий/мл (509 и 16 411 копий/мл). В результате исследования лекарственной устойчивости методом генотипирования, выполненного 2 пациентам с подтвержденным определяемым уровнем РНК ВИЧ-1, у 1 были выявлены многочисленные мутации, обуславливающие резистентность к НИОТ (L74V/I, V75T/M/A/S, K65R/N/E, M184V/I, Q151M) и ННИОТ (K103N/S/H/T/R/Q/E, V106A/M/I, Y188L/C/H/F, G190A/S/E/Q/C/T/V). Остальным 2 пациентам с положительными результатами ВН на Визите 2 повторные анализы не проводили, и они продолжили участие в исследовании (на последующих визитах РНК ВИЧ-1 не выявлялась).
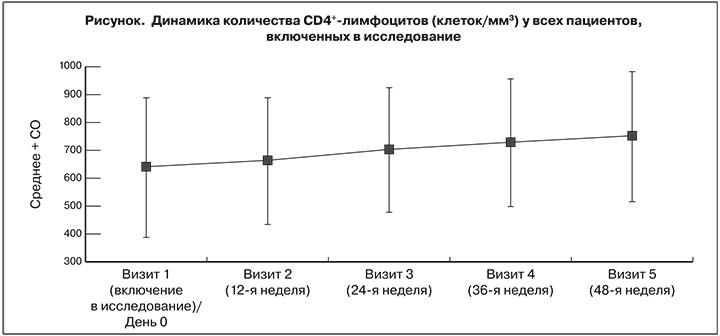
На фоне АРТ по схеме LPV/r + 3TC на 24-й, 36-й и 48-й неделях значительно возросло количество CD4+-лимфоцитов, что подтвердили результаты оценки средних значений и медиан наблюдаемых сдвигов и соответствующие двухсторонние 95% ДИ. Количество CD4+-лимфоцитов на 24-й неделе в среднем увеличились на 64,70 ± 165,06 клеток/мм3 [95% ДИ 41,97–87,43], на 36-й – на 89,34 ± 200,27 [95% ДИ: 61,69–116,98] и на 48-й – на 111,75 ± 184,48 [95% ДИ: 86,15–137,34] (см. рисунок ). Среднее изменение составило соответственно 0,96 ± 4,88 [95% ДИ: 0,28–1,63], 1,28 ± 5,17 [95% ДИ: 0,57–2,00] и 1,43 ± 6,31% [95% ДИ: 0,55–2,31]
Большинство пациентов хорошо переносили терапию по схеме LPV/r + 3TC. Статистически значимых изменений средних значений антропометрических (обхват плеча, бедра, талии) и лабораторных показателей (глюкоза, инсулин, общий холестерин, холестерин, ЛПНП и ЛПВП, триглицериды, креатинин, АЛТ, АСТ) по сравнению с исходным уровнем выявлено не было.
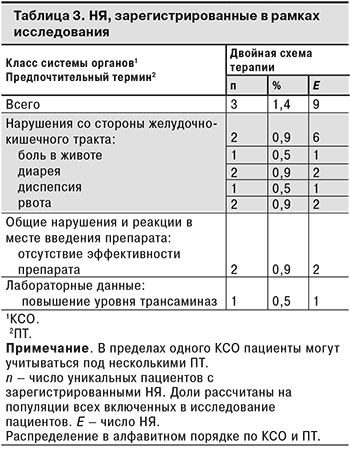
Спонтанные сообщения о НЯ были зарегистрированы всего у 3 (1,4%) пациентов. Поскольку исследование было неинтервенционным, НЯ регистрировали методом спонтанных сообщений в соответствии с требованиями руководства EMA по надлежащей практике фармаконадзора (GVP, модуль VI) и национальной практикой фармаконадзора в Российской Федерации. Большинство НЯ относились к нарушениям со стороны желудочно-кишечного тракта: у 2 (0,9%) пациентов были зарегистрированы 6 НЯ этого класса. Все НЯ были несерьезными и не были связаны с проводимой терапией. Информация о НЯ, зарегистрированных в рамках исследования, представлена в табл. 3.
У 3 (1,4%) пациенток на фоне АВТ LPV/r + 3TC наступила беременность. Хотя все они по этой причине прекратили участие в исследовании, 5 случаев экспозиции во время беременности были зарегистрированы в базе данных как «воздействие терапии на плод во время беременности» и/или «воздействие терапии во время беременности». Сведения об исходе беременности были получены только в 1 случае: она разрешилась рождением живого ребенка (случай зарегистрирован как «живорождение»).
Обсуждение
Результаты настоящего наблюдательного исследования дополняют результаты ранее проведенных клинических исследований OLE [8] и GARDEL [7]. Двойной режим, включающий LPV/r + 3TC в суточных дозах 800/200 мг + 300 мг, может рассматриваться в качестве эффективной и хорошо переносимой упрощенной схемы АРТ для пациентов с достигнутой супрессией ВИЧ. Эти результаты имеют клиническое значение, поскольку свидетельствуют о не меньшей клинической, вирусологической и иммунологической эффективности АРТ при переключении на двойной режим в течение 48 нед. Популяция пациентов, включенных в данное исследование, была сопоставима с популяцией пациентов в исследовании OLE в части основных исходных характеристик и основных (первичных) результатов. Но эти 2 исследования имеют различные результаты по ряду дополнительных (вторичных) показателей эффективности. В исследовании OLE среднее увеличение количества CD4+-лимфоцитов на 48-й неделе по сравнению с исходным уровнем составило 29 ± 182 клеток/мм3 в группе, получавшей двухкомпонентную терапию, и 40 ± 254 клеток/мм3 в группе, получавшей трехкомпонентную терапию, без статистически значимой разницы (p = 0,75) [8]. В настоящем исследовании этот показатель (111,75 ± 184,48 клеток/мм3 на 48-й неделе), по крайней мере, в 2 раза превышал значения, зарегистрированные в исследовании OLE. Хотя динамику изменения количества CD4+-лимфоцитов до начала лечения в рамках данного исследования не регистрировали, такой благоприятный результат может быть связан с прекращением терапии ZDV у большинства участников исследования SIMPLE. Другим значительным отличием является более низкая частота зарегистрированных НЯ в группе пациентов, получавших схему LPV/r + 3TC (1,4% против 53% в исследовании OLE). В настоящем исследовании не было зарегистрировано НЯ, связанных с проводимой терапией, или серьезных НЯ.
Ограничения настоящего исследования включали отсутствие контрольной группы для сравнения вирусологического и иммунологического ответа, а также профиля безопасности с пациентами, продолжающими получать текущую схему терапии. Исследование имело наблюдательный дизайн, в рамках которого бо́льшая часть данных была получена из медицинских карт пациентов и по результатам лабораторных анализов. Качество полученной информации могло варьировать, что можно объяснить влиянием человеческого фактора и возможным пропуском некоторых данных. Нельзя полностью исключить наличие систематических ошибок и их возможное влияние на результаты исследования.
В общей сложности в исследовании приняли участие 216 пациентов из 13 федеральных и республиканских центров, расположенных в 12 городах Российской Федерации. Учитывая относительно небольшую когорту пациентов, эти данные не могут быть экстраполированы на популяцию больных ВИЧ в Российской Федерации в целом. При этом исследуемая выборка характеризуется гомогенностью, так как в ней были представлены пациенты из разных регионов страны.
Заключение
Переключение ВИЧ-1-инфицированных пациентов с опытом АРТ на режим LPV/r + 3TC в условиях рутинной клинической практики в Российской Федерации обеспечивало поддержание вирусологической супрессии, улучшало иммунный ответ, было безопасно и хорошо переносилось в течение 48 нед. лечения.
* * *
Благодарность
Авторы выражают благодарность всем главным исследователям, принимавшим участие в проведении данного исследования.


